

Introduzione
L’articolo cerca di essere il più esaustivo possibile, e parte dalle basi (terminologia, concetti essenziali) per poi approfondire le varie tematiche collegate agli eventi avversi. Se hai già chiari i concetti fondamentali o se il tuo interesse principale è scoprire quali eventi avversi sono stati rilevati per i cosiddetti “vaccini” anti COVID-19, oppure dove trovare la letteratura scientifica in merito, o ancora, come segnalare un evento avverso, usa il sommario qua sotto per visitare soltanto i capitoli che ti interessano.
Sommario

- Definizioni dei termini in medicina
- Alcune premesse sul fenomeno degli eventi avversi
- Autorizzazione condizionata dei medicinali per uso umano: previsioni di legge
- Codice deontologico: doveri del medico e diritti del cittadino
- Farmacovigilanza
- Eventi avversi emersi finora per queste terapie geniche sperimentali
- Informazioni sugli eventi avversi: dove trovarle
- Segnalazione di eventi avversi: procedure
Definizioni dei termini in medicina
 Danno: alterazione, temporanea o permanente, di una parte del corpo o di una funzione fisica o psichica (compresa la percezione del dolore).
Danno: alterazione, temporanea o permanente, di una parte del corpo o di una funzione fisica o psichica (compresa la percezione del dolore).
Effetti collaterali o indesiderati (“side effects”): effetti dovuti al meccanismo d’azione del farmaco e correlati alla dose somministrata. Si manifestano alle normali dosi terapeutiche e si verificano in organi o distretti diversi da quelli desiderati. Sono principalmente dovuti alla distribuzione del farmaco in tutto l’organismo. Non sempre sono nocivi.
Effetti tossici: sono dovuti al meccanismo d’azione del farmaco e correlati alla dose somministrata. Sono espressione della tossicità del farmaco e compaiono a dosi elevate, ma si possono verificare anche a dosi terapeutiche in particolari condizione cliniche.
Errore: fallimento nella pianificazione e/o nell’esecuzione di una sequenza di azioni, che determina il mancato raggiungimento dell’obiettivo desiderato.
Evento (“incident”): Accadimento che ha dato (o aveva la potenzialità di dare) origine a un danno non intenzionale in un paziente.
Evento avverso: è un qualsiasi episodio sfavorevole che si verifica dopo la somministrazione di un farmaco o di un vaccino, ma che non è necessariamente causato dall’assunzione del farmaco o dall’aver ricevuto la vaccinazione e può essere correlato a più variabili. Diversamente si parla di reazione avversa (rappresentata anche con una sigla che è ADR: Adverse Drug Reaction) quando l’evento inatteso correlato al processo assistenziale comporta un danno al paziente, non intenzionale e non desiderato, determinando il nesso di causalità fra farmaco e danno. Il danno può essere prevenibile (quello da ricondurre ad errore) o non prevenibile. Include i danni da farmaci derivanti da:
- Uso non conforme alle indicazioni contenute nell’autorizzazione all’immissione in commercio
- Errori terapeutici, incluso il sovradosaggio accidentale
- Uso improprio
- Abuso del farmaco
- Associazione all’esposizione per motivi professionali
Evento evitato: errore che ha la potenzialità di causare un evento avverso, che tuttavia non si verifica per un caso fortuito, o perché intercettato o perché non ha conseguenze avverse per il paziente.
Evento sentinella: evento avverso di particolare gravità, potenzialmente indicativo di un serio malfunzionamento del sistema, che può comportare morte o grave danno al paziente, e che determina una perdita di fiducia dei cittadini nei confronti del servizio sanitario. Per la particolare gravità, è sufficiente che si verifichi una sola volta perché da parte dell’organizzazione si renda opportuna:
- Un’indagine immediata per accertare quali fattori eliminabili o riducibili lo abbiano causato o vi abbiano contribuito;
- L’implementazione delle adeguate misure correttive.
Farmacosorveglianza: è la disciplina che identifica tutte le attività che vengono svolte per monitorare gli effetti dei farmaci sull’uomo. Le attività di sorveglianza vanno svolte in tutte le fasi della vita di un farmaco, incluso il periodo dello sviluppo clinico. A livello internazionale più correttamente si tende a utilizzare il termine “drug safety surveillance”, specificando se è pre o post-marketing (prima o dopo l’immissione in commercio).
Farmacovigilanza: l’insieme delle attività il cui obiettivo è quello di fornire, in modo continuativo, le migliori informazioni possibili sulla sicurezza dei farmaci, permettendo così di adottare le misure opportune e pertanto assicurare che i farmaci disponibili sul mercato presentino, nelle condizioni di utilizzo autorizzate, un rapporto beneficio-rischio favorevole per la popolazione.
Nel 2002 è stata definita dall’Organizzazione Mondiale della Sanità (OMS) come “la scienza e le attività collegate all’identificazione, valutazione, comprensione e prevenzione delle reazioni avverse o di altri problemi collegati ai farmaci”.
Governo clinico (“clinical governance”): sistema attraverso il quale le organizzazioni sanitarie si rendono responsabili del miglioramento continuo della qualità dei loro servizi e garantiscono elevati standard assistenziali creando le condizioni ottimali nelle quali viene favorita l’eccellenza clinica.
Letalità: tasso che calcola quante persone sono decedute per una determinata malattia espressa.
Mortalità: tasso che calcola quante persone sono decedute sul totale delle persone esposte (cioè gli abitanti di una zona geografica).
Rischio (“risk”): condizione o evento potenziale, intrinseco o estrinseco al processo, che può modificare l’esito atteso del processo. È misurato in termini di probabilità e di conseguenze, come prodotto tra la probabilità che accada uno specifico evento (P) e la gravità del danno che ne consegue (D); nel calcolo del rischio si considera anche la capacità del fattore umano di individuare in anticipo e contenere le conseguenze dell’evento potenzialmente dannoso (fattore K).
Alcune premesse sul fenomeno degli eventi avversi
Il sistema sanitario è un sistema complesso in cui interagiscono molteplici fattori, eterogenei e dinamici, tra cui si citano la pluralità delle prestazioni sanitarie, delle competenze specialistiche e dei ruoli professionali, tecnico-sanitari ed economico-amministrativi e l’eterogeneità dei processi e dei risultati da conseguire. Tutti gli elementi del sistema devono integrarsi e coordinarsi, per rispondere ai bisogni assistenziali del paziente e assicurargli la miglior cura possibile. Nell’ambito sanitario, per la sua complessità, possono verificarsi incidenti ed errori. Rispetto a quanto accade in altri sistemi complessi, in sanità le procedure di sicurezza non possono avere un’impronta prevalentemente “meccanicistica”, in quanto in questo ambito prevale il “fattore umano”, che è nel contempo risorsa e criticità. Vengono pertanto progettati specifici modelli di controllo del rischio clinico, con l’obiettivo di prevenire il verificarsi di un errore e, qualora questo accada, contenerne le conseguenze. La prevenzione dell’errore deve guardare quindi a quello che può essere il fattore “attivo”, ovvero l’errore umano, e a quelli che si definiscono “fattori latenti”, come le insufficienze o errori di progettazione, organizzazione e controllo che restano silenti nel sistema, finché un fattore scatenante non li rende manifesti in tutta la loro potenzialità, causando danni più o meno gravi.
Autorizzazione condizionata dei medicinali per uso umano: previsioni di legge

Le autorizzazioni all’immissione in commercio condizionate si differenziano dalle autorizzazioni all’immissione in commercio rilasciate in circostanze eccezionali conformemente all’articolo 14, paragrafo 8, del regolamento (CE) n. 726/2004 del Parlamento Europeo e del Consiglio. L’autorizzazione all’immissione in commercio condizionata è rilasciata prima che tutti i dati siano disponibili e viene subordinata alla presenza di determinate condizioni e a specifici obblighi, al fine di conseguire il giusto bilanciamento fra l’agevolare l’accesso ai medicinali ai pazienti con necessità mediche insoddisfatte, e l’evitare di autorizzare medicinali che presentano un rapporto rischio/beneficio sfavorevole. Essa non è tuttavia destinata a rimanere condizionata a tempo indeterminato. Quando vengono forniti i dati mancanti dovrebbe piuttosto essere possibile sostituirla con un’autorizzazione all’immissione in commercio non condizionata, vale a dire non subordinata a obblighi specifici.
L’autorizzazione all’immissione in commercio condizionata secondo il Regolamento (CE) n. 507/2006 della Commissione (Commissione delle Comunità Europee), relativo ai medicinali per uso umano che rientrano nel campo d’applicazione del regolamento (CE) n. 726/2004, può essere rilasciata solo quando si ritiene che, malgrado non siano stati forniti dati clinici completi in merito alla sicurezza e all’efficacia del medicinale, siano rispettate tutte le seguenti condizioni:
a) il rapporto rischio/beneficio del medicinale risulti a favore del beneficio;
b) è probabile che il richiedente possa in seguito fornire dati clinici completi;
c) il medicinale risponda ad esigenze mediche insoddisfatte;
d) i benefici per la salute pubblica derivanti dalla disponibilità immediata sul mercato del medicinale in questione superino il rischio inerente al fatto che occorrano ancora dati supplementari.
In particolare il punto A, cioè la valutazione del rapporto rischio/beneficio, è un aspetto fondamentale di ogni farmaco e terapia, non solo sperimentale, ed ha rilievo non solo scientifico ma anche etico e giuridico; e ciò a maggior ragione se si tratta di un farmaco somministrato a soggetti sani al fine di prevenire il rischio di sviluppare una malattia in forma grave: come sottolineato dall’AIFA, poiché in ambito vaccinale la popolazione target è rappresentata prevalentemente da soggetti sani, il livello accettabile di rischio è inferiore a quello degli altri prodotti medicinali specialmente con riguardo alla popolazione pediatrica.
Tuttavia, anche il punto C (esigenze mediche insoddisfatte), è di grande importanza, e non è stato rispettato. Come è ormai ben noto, sia dalle esperienze extranazionali ed extraeuropee, sia da quelle nazionali, sono state usate e continuano a essere usate molteplici terapie per la cura del COVID-19, molte delle quali di comprovata efficacia.
Codice deontologico: doveri del medico e diritti del cittadino
 In questa sezione si affrontano le tematiche di: rapporto rischio/beneficio, principio di precauzione, primum non nocere, informazioni esaustive al fine di ricevere il consenso dalla persona.
In questa sezione si affrontano le tematiche di: rapporto rischio/beneficio, principio di precauzione, primum non nocere, informazioni esaustive al fine di ricevere il consenso dalla persona.
Il codice della deontologia medica comprende tra gli altri, tre articoli di rilievo:
Art 14 (Prevenzione e gestione di eventi avversi e sicurezza delle cure): “Il medico opera al fine di garantire le più idonee condizioni di sicurezza del paziente e degli operatori coinvolti, promuovendo a tale scopo l’adeguamento dell’organizzazione delle attività e dei comportamenti professionali e contribuendo alla prevenzione e alla gestione del rischio clinico attraverso:
– l’adesione alle buone pratiche cliniche;
– l’attenzione al processo di informazione e di raccolta del consenso, nonché alla comunicazione di un evento indesiderato e delle sue cause;
– lo sviluppo continuo di attività formative e valutative sulle procedure di sicurezza delle cure;
– la rilevazione, la segnalazione e la valutazione di eventi sentinella, errori, “quasi-errori” ed eventi avversi valutando le cause e garantendo la natura riservata e confidenziale delle informazioni raccolte.”
Questo articolo è una traduzione in chiave moderna del principio ippocratico ”primum non nocere” (per prima cosa, non nuocere).

Art.33 (informazione e comunicazione con la persona assistita): “Il medico garantisce alla persona assistita o al suo rappresentante legale un’informazione comprensibile ed esaustiva sulla prevenzione, sul percorso diagnostico, sulla diagnosi, sulla prognosi, sulla terapia e sulle eventuali alternative diagnostico-terapeutiche, sui prevedibili rischi e complicanze, nonché sui comportamenti che il paziente dovrà osservare nel processo di cura.
Il medico adegua la comunicazione alla capacità di comprensione della persona assistita o del suo rappresentante legale, corrispondendo a ogni richiesta di chiarimento, tenendo conto della sensibilità e reattività emotiva dei medesimi, in particolare in caso di prognosi gravi o infauste, senza escludere elementi di speranza.
Il medico rispetta la necessaria riservatezza dell’informazione e la volontà della persona assistita di non essere informata o di delegare ad altro soggetto l’informazione, riportandola nella documentazione sanitaria.
Il medico garantisce al minore elementi di informazione utili perché comprenda la sua condizione di salute e gli interventi diagnostico-terapeutici programmati, al fine di coinvolgerlo nel processo decisionale.”
Questo articolo si ricollega a quanto stabilisce il Comitato Nazionale di Bioetica che guardando al rispetto del soggetto, valuta l’informazione come non finalizzata a colmare l’inevitabile differenza di conoscenze tecniche tra medico e paziente, ma a porre lo stesso paziente nella condizione di esercitare correttamente i suoi diritti e quindi di formarsi una volontà che sia effettivamente tale, in altri termini in condizioni di scegliere.

Art. 48 (sperimentazione umana): “Il medico attua sull’uomo le sperimentazioni sostenute da protocolli scientificamente fondati e ispirati al principio di salvaguardia della vita e dell’integrità psico-fisica e nel rispetto della dignità della persona.
La sperimentazione sull’uomo è subordinata al consenso informato scritto del soggetto reclutato e alla contestuale e idonea informazione del medico curante indicato dallo stesso.
Il medico informa il soggetto reclutato in merito agli scopi, ai metodi, ai benefici prevedibili e ai rischi, fermo restando il diritto dello stesso di interrompere la sperimentazione in qualsiasi momento, garantendo in ogni caso la continuità assistenziale.
Nel caso di minore o di persona incapace, la sperimentazione è ammessa solo per finalità preventive o terapeutiche relative alla condizione patologica in essere o alla sua evoluzione.
Il medico documenta la volontà del minore e ne tiene conto.”
Questo articolo si presta alle riflessioni deontologiche sulle conclusioni del Processo di Norimberga (1946-1947) che condannò i medici nazisti per gli esperimenti criminali compiuti sui prigionieri nei campi di sterminio. In seguito a questi fatti, per la prima volta nella storia, una Corte di Giustizia riconobbe l’ammissibilità della sperimentazione sull’uomo, purché rispettosa dei suoi diritti, e indicò nel Decalogo di Norimberga i principi etici e giuridici secondo i quali perseguire scienziati e ricercatori responsabili di sperimentazioni criminali sulle persone. Quindi in una sperimentazione non si può prescindere da questi elementi:
- Consenso libero e informato da parte di chi si sottopone alla sperimentazione.
- Chi conduce la sperimentazione è personalmente responsabile della sua validità scientifica.
- Una sperimentazione è giustificata sulla base dei risultati che ci si attende di ottenere; il livello del rischio non deve mai superare il livello di importanza che ha il problema da risolvere mediante la sperimentazione.
Il medico ricercatore dunque è tenuto a rispettare il Codice deontologico medico, le Direttive di Ginevra, la Dichiarazione di Helsinki, la Convenzione di Oviedo, la dichiarazione universale di Bioetica e dei diritti umani (Unesco).
Farmacovigilanza
Le segnalazioni di sospetta reazione avversa sono inserite nella Rete Nazionale di Farmacovigilanza (RNF) per ogni vaccino in relazione al numero di dosi somministrate, alla distribuzione per sesso e fascia di età.
Vigilanza passiva
Raccolta dei dati che derivano da comunicazioni spontanee non sollecitate provenienti da parte di un osservatore (paziente o segnalatore identificabile: medico, infermiere, farmacista, cittadini o paziente medesimo) in merito a una reazione che si sospetta essere insorta a seguito dell’assunzione di un farmaco. La segnalazione viene poi valutata da una speciale commissione medica.
I limiti di questa vigilanza sono essenzialmente legati alla mancata informazione al cittadino su possibilità e modalità di segnalazione e alla minimizzazione da parte del sanitario in merito alla necessità di segnalazione che dunque spesso viene omessa e di fatto il medico non è soggetto a sanzione nonostante quanto sancito dal Decreto Ministeriale del 30 aprile 2015. Con tale decreto l’Italia ha recepito le direttive europee in materia, stabilendo l’obbligo di segnalare tempestivamente le sospette reazioni avverse da farmaci e da vaccini, oltre ad aver definito dei limiti di tempo entro cui gli operatori sanitari sono tenuti ad effettuare la segnalazione alla Rete Nazionale di Farmacovigilanza (RNF) dell’AIFA.
Segnalazione stimolata
La segnalazione stimolata consiste in tutti quei metodi che hanno lo scopo di incoraggiare e facilitare le segnalazioni di sospette reazioni avverse al farmaco da parte degli operatori sanitari e dei cittadini, in ambienti specifici come ospedali, farmacie territoriali, etc. Viene usata principalmente per nuovi farmaci e per periodi di tempo limitati.
Sorveglianza attiva
Si tratta di uno dei compiti che la legge ha affidato ad AIFA, infatti, consiste nel promuovere e coordinare, anche in collaborazione con l’Istituto Superiore di Sanità, studi e ricerche di “farmacovigilanza attiva”. Prevede che alcune persone che assumono un determinato farmaco vengano seguite per un certo periodo di tempo e siano attentamente monitorate.
Si è visto che, quando ciò accade, le reazioni avverse che vengono annotate sono molte di più di quelle che arrivano alla Rete Nazionale attraverso le segnalazioni spontanee.
La sorveglianza attiva può essere realizzata anche attraverso la revisione delle cartelle cliniche o intervistando i pazienti e/o i medici in “siti sentinella”, in modo da garantire dati completi e accurati sugli eventi avversi. La sorveglianza in siti selezionati può fornire diverse informazioni, come i dati provenienti da specifici sottogruppi di pazienti, che non potrebbero essere ottenuti attraverso un sistema di segnalazione spontanea passivo. Tra i maggiori limiti dei siti sentinella vi sono i problemi legati al bias di selezione, il piccolo numero di pazienti e i costi elevati. La sorveglianza attiva è più efficiente per quei farmaci usati principalmente negli ambienti istituzionali come gli ospedali, le case di cura, i centri di emodialisi, etc. Gli ambienti istituzionali possono avere una maggiore frequenza d’uso per certi prodotti farmaceutici e possono disporre di una struttura da dedicare alla segnalazione. Il monitoraggio intensivo da parte dei siti sentinella può, inoltre, essere vantaggioso nell’identificazione dei rischi tra i pazienti.
Monitoraggio intensivo (drug event monitoring)
È un metodo di sorveglianza attiva nel quale i pazienti possono essere identificati da dati di prescrizione elettronici o da sistemi automatizzati a disposizione delle strutture sanitarie. Può inoltre essere spedito un questionario a ogni medico prescrittore o a ogni paziente a intervalli predeterminati, in modo che si possano ottenere ulteriori informazioni sugli esiti dell’evento. I limiti del Drug Event Monitoring includono le basse percentuali di risposta da parte dei medici e dei pazienti e la natura imprecisata della fonte da cui provengono i dati. Inoltre, il mantenimento della privacy del paziente può essere un problema. D’altro canto, possono essere raccolte delle informazioni più dettagliate sugli eventi avversi provenienti da un ampio numero di medici e di pazienti.
Utilizzo dei Registri
Un registro è una lista di pazienti che presentano le stesse caratteristiche; questa caratteristica può essere una patologia (registro di patologia) o una specifica esposizione (registro di farmaco). Entrambe le tipologie di registro, che differiscono solo per i dati di interesse sui pazienti, possono permettere la raccolta di una serie di informazioni, usando questionari standardizzati in maniera prospettica. I registri di patologia possono aiutare la raccolta di dati sull’assunzione dei farmaci e di altri fattori associati alla condizione clinica. Un registro di patologia può, inoltre, essere usato come base per uno studio caso controllo che confronti l’esposizione al farmaco dei casi identificati dal registro e i controlli selezionati, sia da pazienti con altre condizioni, presenti nel registro, sia da pazienti non presenti nel registro. I registri d’esposizione ai farmaci riportano, invece, le popolazioni esposte ai farmaci d’interesse, per determinare se un farmaco ha un impatto speciale su questo gruppo di pazienti. I pazienti presenti in questi registri possono essere seguiti nel tempo ed essere inclusi in uno studio di coorte, per raccogliere dati sugli eventi avversi usando questionari standardizzati.
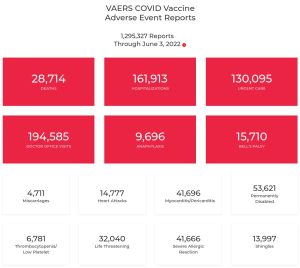
Eventi avversi emersi finora per queste terapie geniche sperimentali

Dai documenti rilasciati da Pfizer su decisione di un giudice federale americano, oppostosi alla volontà della stessa casa farmaceutica di conservare segreti i contratti per ben 75 anni, si evince una lunga serie di eventi avversi, fra i quali citiamo (in ordine alfabetico):
- Amiloidosi gastrointestinale
- Anticorpo anti-sperma positivo
- Autoimmunità testicolare
- Basso peso alla nascita
- Crisi tonico-cloniche generalizzate
- Danno al fegato
- Danno renale acuto
- Decesso
- Diabete mellito di tipo 1
- Embolia del tronco cerebrale
- Embolia della vena giugulare
- Encefalite emorragica
- Encefalite tronco encefalico
- Encefalopatia di Hashimoto
- Epatite immuno-mediata
- Epilessia del lobo frontale
- Epilessia del lobo temporale
- Epilessia mioclonica giovanile
- Infarto
- Infarto cerebrale trombotico
- Insufficienza cardiaca
- Malattia polmonare interstiziale
- Mielite flaccida acuta
- Miocardite, crisi neonatale
- Morte neonatale
- Natimortalità
- Pancreatite, polmonite
- Paralisi facciale
- Psicosi epilettica
- Riattivazione dell’herpes zoster (talora con necrosi retinica acuta)
- Schiuma in bocca
- Shock cardiogenico
- Sindrome da sofferenza fetale
- Sindrome infiammatoria multisistemica nei bambini
- Tachicardia
- Trombosi del tronco cerebrale
- Trombosi dell’arteria vertebrale
- Trombosi vascolare epatica
- Trombosi venosa neonatale
- Trombosi venosa profonda
- Trombosi ventricolare cardiaca
- Vasculite del sistema nervoso centrale
Qui trovate l’analisi cumulativa delle segnalazioni di eventi avversi post-autorizzazione della Pfizer.
Tutti i documenti di Pfizer sono scaricabili ai link: https://www.icandecide.org/pfizer/ e https://phmpt.org/pfizers-documents/
Miocardite
Se cercate informazioni specifiche su questo specifico (e piuttosto ricorrente) evento avverso, ecco una raccolta di pubblicazioni e articoli a tema:
➡️ European Journal of Clinical Investigation | 14/02/2022
➡️ Jama Pediatrics | 25/02/2022
➡️ La Verità | 16/02/2022
➡️ Journal of Personalized Medicine | 28/10/2021
➡️ MDPI – Pediatric Reports | 01/09/2021
➡️ Journal of the American Medical Association | 04/08/2021
➡️ MedRxiv | 03/03/2022 (pre-print)
➡️ EMA – Risk Management Plan | 15/12/2021
➡️ Journal of the American Medical Association | 25/01/2022
➡️ Cureus | 03/11/2021
➡️ SpringerLink – Clinical Research in Cardiology | 23/11/2021
➡️ FDA – Pfizer | 26/10/2021 (VRBPAC Briefing Document)
➡️ Global Cardiology Science & Practice | 30/09/2021
➡️ Journal of the American Medical Association – Cardiology | 10/08/2021
➡️ The Lancet | 29/10/2021
➡️ ScienceDirect – Vaccine | 04/09/2021
➡️ QJM: An International Journal of Medicine | 29/09/2021
➡️ QJM: An International Journal of Medicine | 21/09/2021 (Case report)
➡️ Journal of Korean Medical Science | 10/07/2021
➡️ The New England Journal of Medicine | 16/09/2021
➡️ The New England Journal of Medicine | 06/10/2021
➡️ The New England Journal of Medicine | 06/10/2021
➡️ ScienceDirect – Current Problems in Cardiology | 01/10/2021 (Pre-proof | RITIRATO)
➡️ American Heart Association – Circulation | 20/07/2021
➡️ The Lancet – Viewpoint | 13/09/2021
➡️ MedRxiv | 18/08/2021 (Pre-print)
➡️ Pediatrics | 01/09/2021
➡️ Journal of the American Medical Association | 29/06/2021
➡️ MedRxiv | 16/09/2021 (Pre-print | RITIRATO)
➡️ MedRxiv | 08/09/2021 (Pre-print)
➡️ Journal of the American Medical Association | 04/08/2021
Informazioni sugli eventi avversi: dove trovarle
- AIFA (Agenzia Italiana del Farmaco)
- EMA (European Medicines Agency): https://www.ema.europa.eu/en e https://european-union.europa.eu/institutions-law-budget/institutions-and-bodies/institutions-and-bodies-profiles/ema_it
- EUDRAVIGILANCE (database EMA per le reazioni avverse): https://www.adrreports.eu/it/index.html e https://www.adrreports.eu/it/covid19_message.html
- OMS VIGIBASE
- EUROMOMO (per i dati sulla mortalità in Europa)
- VIGIACCESS
- VAERS: (Vaccine Adverse Event Reporting System): https://openvaers.com/covid-data e vaers.hhs.gov e https://www.cdc.gov/coronavirus/2019-ncov/index.html
- CMSI (Commissione Medico Scientifica Indipendente)
- ASSIS
- CONTIAMOCI
- C15O (Coordinamento 15 Ottobre)
- Mille Medici Per La Costituzione: sito web, email e canale Telegram
- Database Italia: sito web e canale Telegram
- Eventi avversi da vaccino Covid: sito web e canale Telegram
- Dr. John B (in lingua inglese): canale Telegram
- Studi scientifici VACCINI COVID-19 SARSCOV-2: canale Telegram
- Umanità e ragione: sito web e canale Telegram
- Lucca Consapevole, progetto Voci Avverse
Segnalazione di eventi avversi: procedure
 La segnalazione di una reazione indesiderata a un farmaco è necessaria per raccogliere e analizzare tutti i dati che servono a conoscere sempre meglio il rapporto beneficio/rischio del farmaco. Il sistema della Farmacovigilanza in Italia è affidato alle segnalazioni spontanee di qualunque figura, sanitaria o no, che venga a conoscenza di un evento avverso.
La segnalazione di una reazione indesiderata a un farmaco è necessaria per raccogliere e analizzare tutti i dati che servono a conoscere sempre meglio il rapporto beneficio/rischio del farmaco. Il sistema della Farmacovigilanza in Italia è affidato alle segnalazioni spontanee di qualunque figura, sanitaria o no, che venga a conoscenza di un evento avverso.
Il cittadino può autonomamente segnalare un evento avverso. Nessuno, sanitario o non sanitario, può essere legittimato a definire a prescindere dalla segnalazione se la reazione avversa è correlata alla assunzione del vaccino oppure no; pertanto nessuno può disincentivare o addirittura impedire la segnalazione medesima che andrà poi al vaglio di apposita commissione di studio.
Opportuno sottolineare che il personale sanitario deve effettuare segnalazione. Il Decreto del Ministero della Salute 30 aprile 2015 ha ribadito l’obbligo di segnalare tempestivamente le sospette reazioni avverse da farmaci e da vaccini e ha definito dei limiti di tempo entro cui gli operatori sanitari sono tenuti ad effettuare la segnalazione alla Rete Nazionale di Farmacovigilanza (RNF) dell’AIFA.
Quali informazioni si devono fornire?
Questi i dati da fornire:
- Caratteristiche, durata ed esito della reazione avversa.
- Iniziali del nome e cognome del soggetto, genere (M/F), età, alcuni dati antropometrici del paziente (peso, altezza, ultimo ciclo mestruale, gravidanza, allattamento, etc.)
- Se possibile, il lotto a cui appartiene il vaccino somministrato, ma l’assenza di questa informazione non compromette la procedura della segnalazione
La segnalazione non è anonima ma tiene nell’anonimato il paziente.
Le segnalazioni sono sottoposte a un algoritmo specifico messo a punto dal Comitato Consultivo Globale per la Sicurezza dei Vaccini (GACVS) dell’Organizzazione Mondiale della Sanità che tiene conto di:
- relazione temporale fra la vaccinazione e la reazione segnalata;
- presenza di possibili spiegazioni alternative;
- prove a favore dell’associazione tra la vaccinazione e la reazione;
- precedenti evidenze di letteratura;
- frequenza dell’evento segnalato nella popolazione generale, anche non vaccinata;
- plausibilità biologica.
Il risultato sarà inquadrato come:
- correlabile: l’associazione causale fra evento e vaccino è considerata plausibile;
- non correlabile: altri fattori possono giustificare l’evento;
- indeterminata: l’associazione temporale è compatibile, ma le prove non sono sufficienti a supportare un nesso di causalità;
- non classificabile: le eventuali segnalazioni prive di informazioni sufficienti rendono necessari ulteriori approfondimenti.
I rapporti AIFA sulla sorveglianza dei vaccini COVID-19, un tempo mensili, ora a cadenza trimestrale, sono consultabili a questo link.
Procedure di segnalazione
Il cittadino, in autonomia o per conto di un sanitario o altro soggetto, ha due strumenti alternativi per inviare segnalazione:
- Telematica diretta: andando sul sito https://www.vigifarmaco.it/ e seguendo le indicazioni richieste. Dal 20 Giugno 2022 il sito sarà convertito con la nuova Rete Nazionale di Farmacovigilanza (NRNF) ed includerà il nuovo modulo web per la segnalazione online.
- Cartacea: ci sono due documenti da scaricare (DOCUMENTO1 —– DOCUMENTO2), compilare e inviare Per FAX o E-MAIL o POSTA al Responsabile di Farmacovigilanza della propria ASL. Gli indirizzi sono presenti sul sito dell’AIFA.
In alternativa si può ricorrere ad alcune realtà che si sono messe a disposizione per aiutare i cittadini nelle segnalazioni. Fra queste citiamo:
- Libertà Livorno, contattandoci tramite il nostro canale Telegram, o scrivendoci un’email con i tuoi contatti a info@libertalivorno.it
- Lucca Consapevole
- M.I.A.S. (Movimento per l’indipendenza e l’autonomia della Sicilia)
- ComeDonChisciotte.org: sito web e email
- Il Filo di Arianna: canale Telegram

 già Libertà Livorno e ciò per cui si batte, o comunque sei interessato esclusivamente ai contenuti di approfondimento riguardo gli eventi avversi (in particolare legati alla terapia genica anti SARS-COV2? Se la risposta è sì, puoi saltare questa sezione e consultare l’articolo dedicato
già Libertà Livorno e ciò per cui si batte, o comunque sei interessato esclusivamente ai contenuti di approfondimento riguardo gli eventi avversi (in particolare legati alla terapia genica anti SARS-COV2? Se la risposta è sì, puoi saltare questa sezione e consultare l’articolo dedicato